Phylogenetic Entropy of a community
PhyloEntropy.RdCalculates the phylogenetic entropy of order \(q\) of a probability vector.
Usage
PhyloEntropy(NorP, q = 1, Tree, Normalize = TRUE, ...)
bcPhyloEntropy(Ns, q = 1, Tree, Normalize = TRUE, Correction = "Best",
SampleCoverage = NULL, CheckArguments = TRUE)
# S3 method for class 'ProbaVector'
PhyloEntropy(NorP, q = 1, Tree, Normalize = TRUE,
..., CheckArguments = TRUE, Ps = NULL)
# S3 method for class 'AbdVector'
PhyloEntropy(NorP, q = 1, Tree, Normalize = TRUE, Correction = "Best",
..., CheckArguments = TRUE, Ns = NULL)
# S3 method for class 'integer'
PhyloEntropy(NorP, q = 1, Tree, Normalize = TRUE, Correction = "Best",
..., CheckArguments = TRUE, Ns = NULL)
# S3 method for class 'numeric'
PhyloEntropy(NorP, q = 1, Tree, Normalize = TRUE, Correction = "Best",
..., CheckArguments = TRUE, Ps = NULL, Ns = NULL)
is.PhyloEntropy(x)
# S3 method for class 'PhyloEntropy'
summary(object, ...)Arguments
- Ps
A probability vector, summing to 1.
- Ns
A numeric vector containing species abundances.
- NorP
A numeric vector, an integer vector, an abundance vector (
AbdVector) or a probability vector (ProbaVector). Contains either abundances or probabilities.- q
A number: the order of entropy. Default is 1.
- Tree
An object of class
hclust, "phylo" (seeread.tree),phylogorPPtree. The tree must be ultrametric.- Normalize
If
TRUE(default), theTotalentropy returned by the function is normalized by the height of the tree (it is the weighted average value of the entropy in each slice).
IfFALSE, it is the unnormalized weighted sum of the results.- Correction
A string containing one of the possible corrections supported by
Tsallis.- SampleCoverage
The sample coverage of
Nscalculated elsewhere. Used to calculate the gamma diversity of meta-communities, see details.- CheckArguments
Logical; if
TRUE, the function arguments are verified. Should be set toFALSEto save time when the arguments have been checked elsewhere.- x
An object to be tested or plotted
- object
A
PhyloEntropyobject to be summarized.- ...
Additional arguments to be passed to the generic methods.
Details
The phylogenetic entropy is its generalization of HCDT entropy to unequal species distances (Pavoine et al., 2009).
Calculation relies on Tsallis and PhyloApply.
Intervals separate two cuts in a tree: no node is found at heights contained in an interval.
Bias correction requires the number of individuals to estimate sample Coverage. Use bcPhyloEntropy and choose the Correction.
The functions are designed to be used as simply as possible. PhyloEntropy is a generic method. If its first argument is an abundance vector, an integer vector or a numeric vector which does not sum to 1, the bias corrected function bcPhyloEntropy is called. Explicit calls to bcPhyloEntropy (with bias correction) or to PhyloEntropy.ProbaVector (without correction) are possible to avoid ambiguity. The .integer and .numeric methods accept Ps or Ns arguments instead of NorP for backward compatibility.
The size of a metacommunity (see MetaCommunity) is unknown so it has to be set according to a rule which does not ensure that its abundances are integer values. Then, classical bias-correction methods do not apply. Providing the SampleCoverage argument allows applying the "ChaoShen" and "Grassberger" corrections to estimate quite well the entropy. DivPart and GammaEntropy functions use this tweak.
Note
The tree must contain all species of the probability vector. If it contains extra species, computation time will just be increased.
Value
An object of class PhyloEntropy is a list:
- Distribution
The distribution used to calculate entropy
- Function
The function used to calculate entropy
- Tree
The functional or phylogenetic tree used to calculate entropy
- Normalized
Logical. Indicates whether phyloentropy is normalized or proportional to the height of the tree.
- Type
The type of entropy ("alpha", "beta" or "gamma").
- Order
The order of entropy \(q\).
- Cuts
A named vector containing values of neutral entropy along the tree. Names are cut ends, i.e. the ends of intervals (the first interval starts at 0 for leaves, the max value is the height of the tree).
- Total
A value equal the total entropy multiplied by the tree height if
Normalizeis FALSE.
is.PhyloEntropy returns TRUE if the object is of class PhyloEntropy.
summary.PhyloEntropy returns a summary of the object's value.
PhyloEntropy objects can be plotted by plot.PhyloValue because PhyloEntropy objects are also of class PhyloValue.
References
Marcon, E., Herault, B. (2015). Decomposing Phylodiversity. Methods in Ecology and Evolution 6(3): 333-339.
Pavoine, S., Love, M. S. and Bonsall, M. B. (2009). Hierarchical partitioning of evolutionary and ecological patterns in the organization of phylogenetically-structured species assemblages: Application to rockfish (genus: Sebastes) in the Southern California Bight. Ecology Letters 12(9): 898-908.
Examples
# Load Paracou data (number of trees per species in two 1-ha plot of a tropical forest
# and their taxonomy)
data(Paracou618)
# Ps is the vector of probabilities
Ps <- as.ProbaVector(Paracou618.MC$Ps)
# Calculate the phylogenetic Shannon entropy of the plot
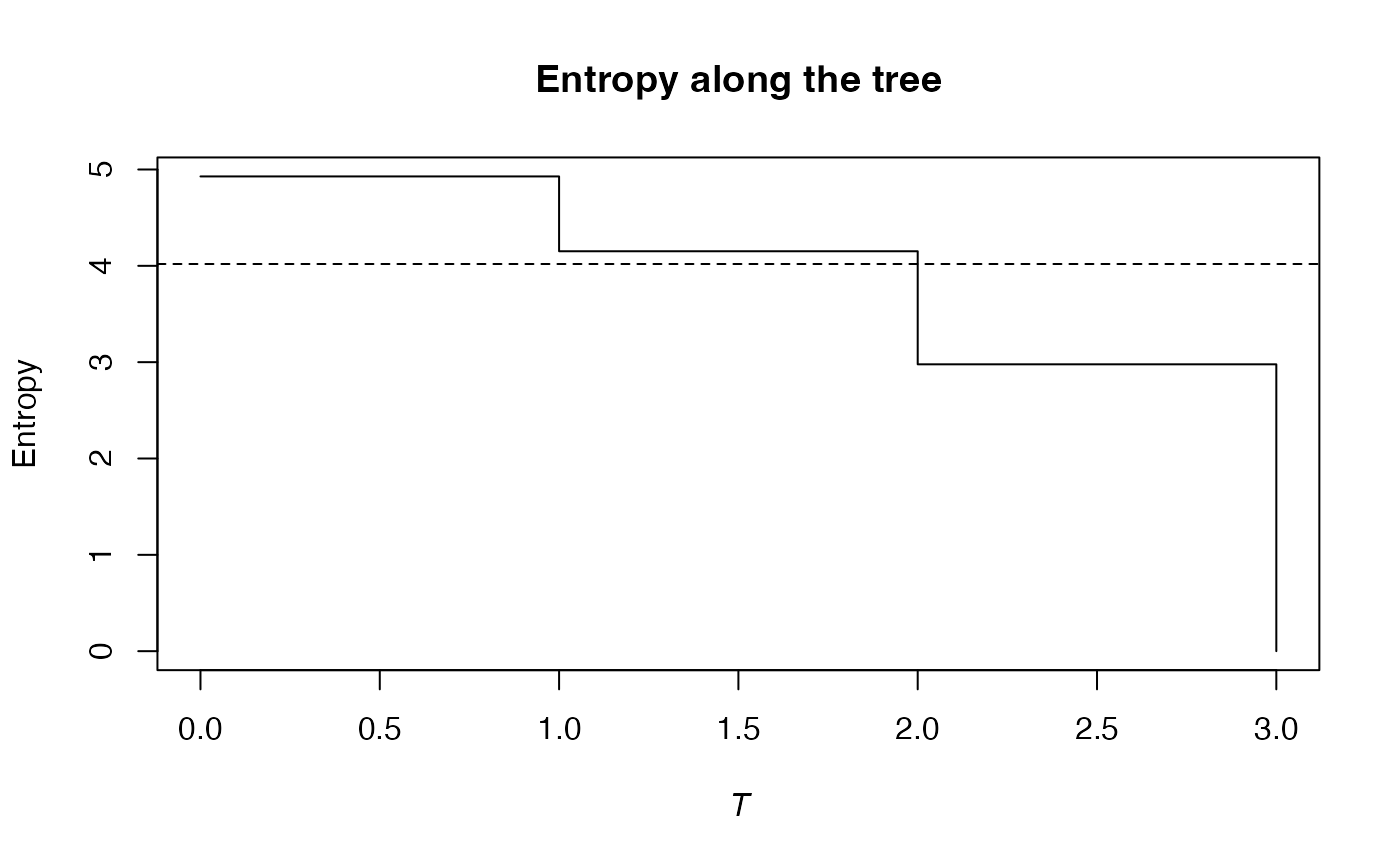
summary(PhyloEntropy(Ps, 1, Paracou618.Taxonomy) -> e)
#> alpha or gamma phylogenetic or functional entropy of order 1 of distribution -
#> with correction: None
#> Phylogenetic or functional entropy was calculated according to the tree
#> Paracou618.Taxonomy
#>
#> Entropy is normalized
#>
#> Entropy equals: 3.919941
plot(e)
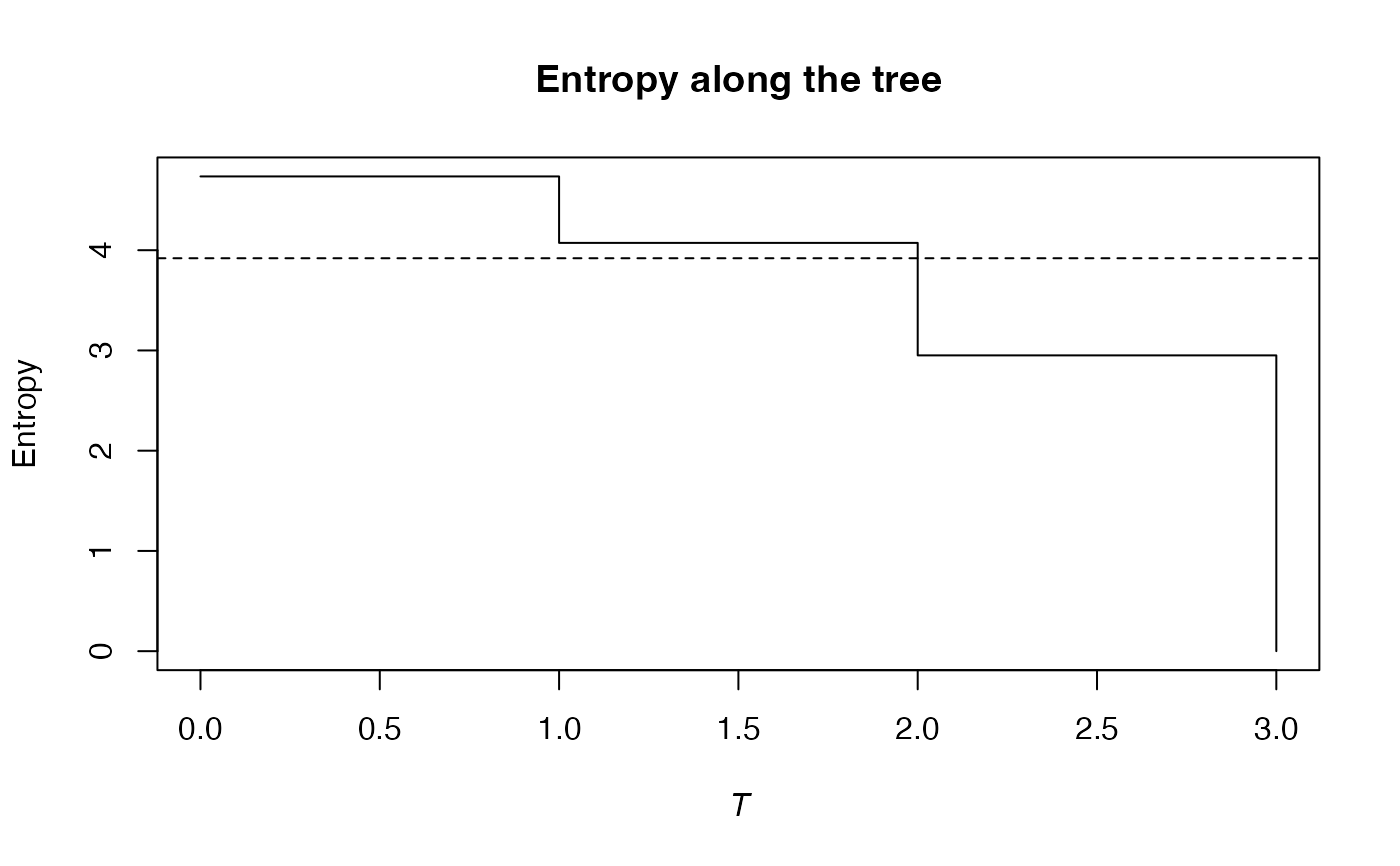 # Ns is the vector of abundances of the metacommunity
Ns <- as.AbdVector(Paracou618.MC$Ns)
# Calculate the phylogenetic Shannon entropy of the plot
summary(bcPhyloEntropy(Ns, 1, Paracou618.Taxonomy, Correction = "Best") -> e)
#> alpha or gamma phylogenetic or functional entropy of order 1 of distribution -
#> with correction: Best
#> Phylogenetic or functional entropy was calculated according to the tree
#> Paracou618.Taxonomy
#>
#> Entropy is normalized
#>
#> Entropy equals: 4.018993
plot(e)
# Ns is the vector of abundances of the metacommunity
Ns <- as.AbdVector(Paracou618.MC$Ns)
# Calculate the phylogenetic Shannon entropy of the plot
summary(bcPhyloEntropy(Ns, 1, Paracou618.Taxonomy, Correction = "Best") -> e)
#> alpha or gamma phylogenetic or functional entropy of order 1 of distribution -
#> with correction: Best
#> Phylogenetic or functional entropy was calculated according to the tree
#> Paracou618.Taxonomy
#>
#> Entropy is normalized
#>
#> Entropy equals: 4.018993
plot(e)