entropart is a package for R designed to estimate diversity based on HCDT entropy or similarity-based entropy.
Several object classes can be used in entropart to represent phylogenies and calculate phylogenetic diversity. They are detailed here.
Formats used
Supported phylogenies are classes phylo from package
ape, phylog from package ade4 and
hclust from package base.
To optimize performance, a specific class PPtree, for
“preprocessed trees”, is introduced by the package: it is basically a
list containing a phylo and an hclust
representation of the phylogeny and preprocessed useful statistics.
The arguments Tree or PhyloTree used in
phylogenetic diversity estimation functions of the package may be any of
those formats.
Conversion between classes
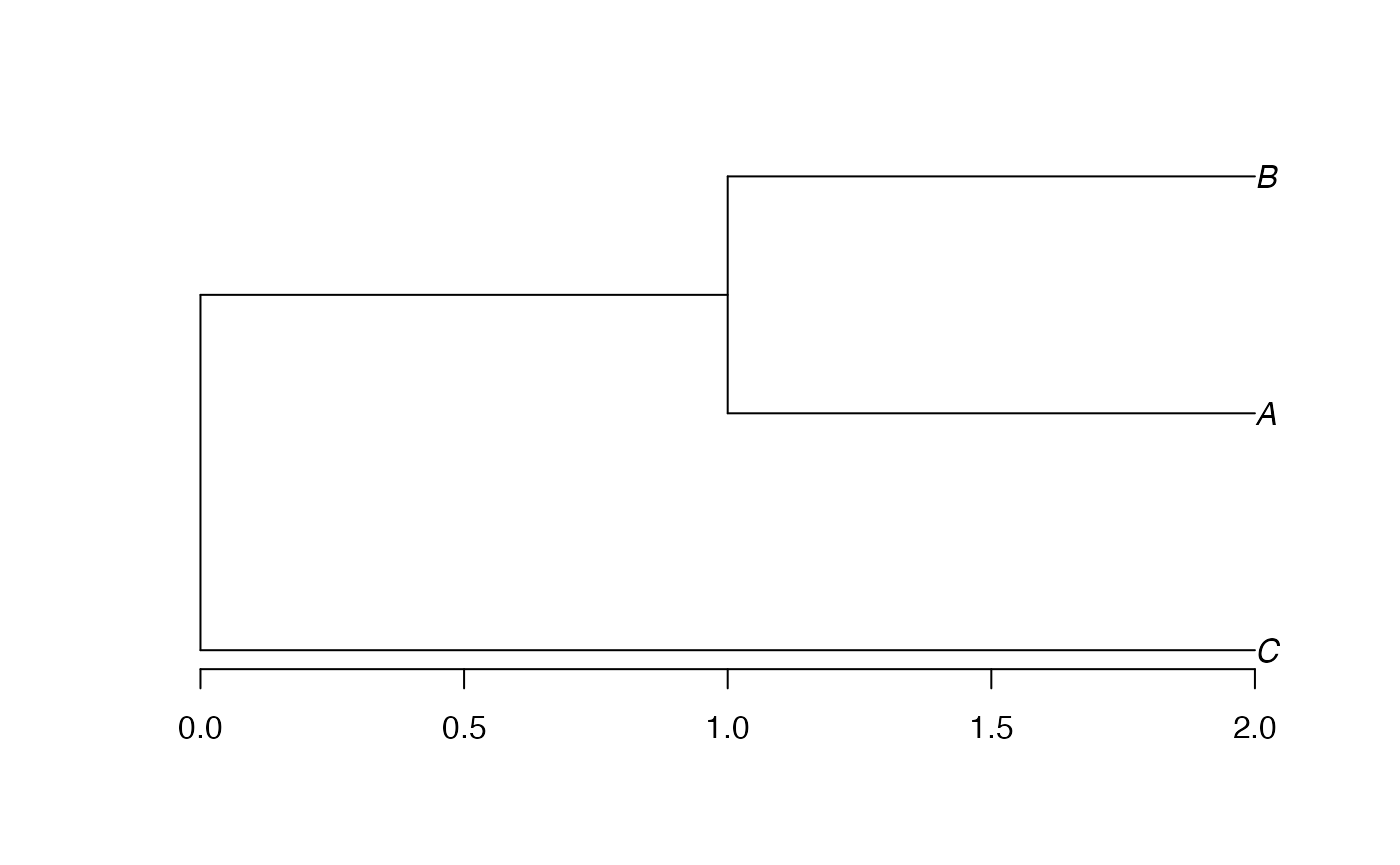
Let’s start from an ultrametric distance matrix. Three species are defined, with distance 1 between the first 2 and distance 2 between both and the last one.
dSp <- matrix(c(0, 1, 2, 1, 0, 2, 2, 2, 0), nrow=3, byrow=TRUE)
row.names(dSp) <- colnames(dSp) <- c("A", "B", "C")
dSp## A B C
## A 0 1 2
## B 1 0 2
## C 2 2 0hclust
An hclust object is created by UPGMA hierarchical
clustering.
require("stats")
plot(hTree <- hclust(as.dist(dSp), method="average"), hang=-0.01, axes = F)
axis(2)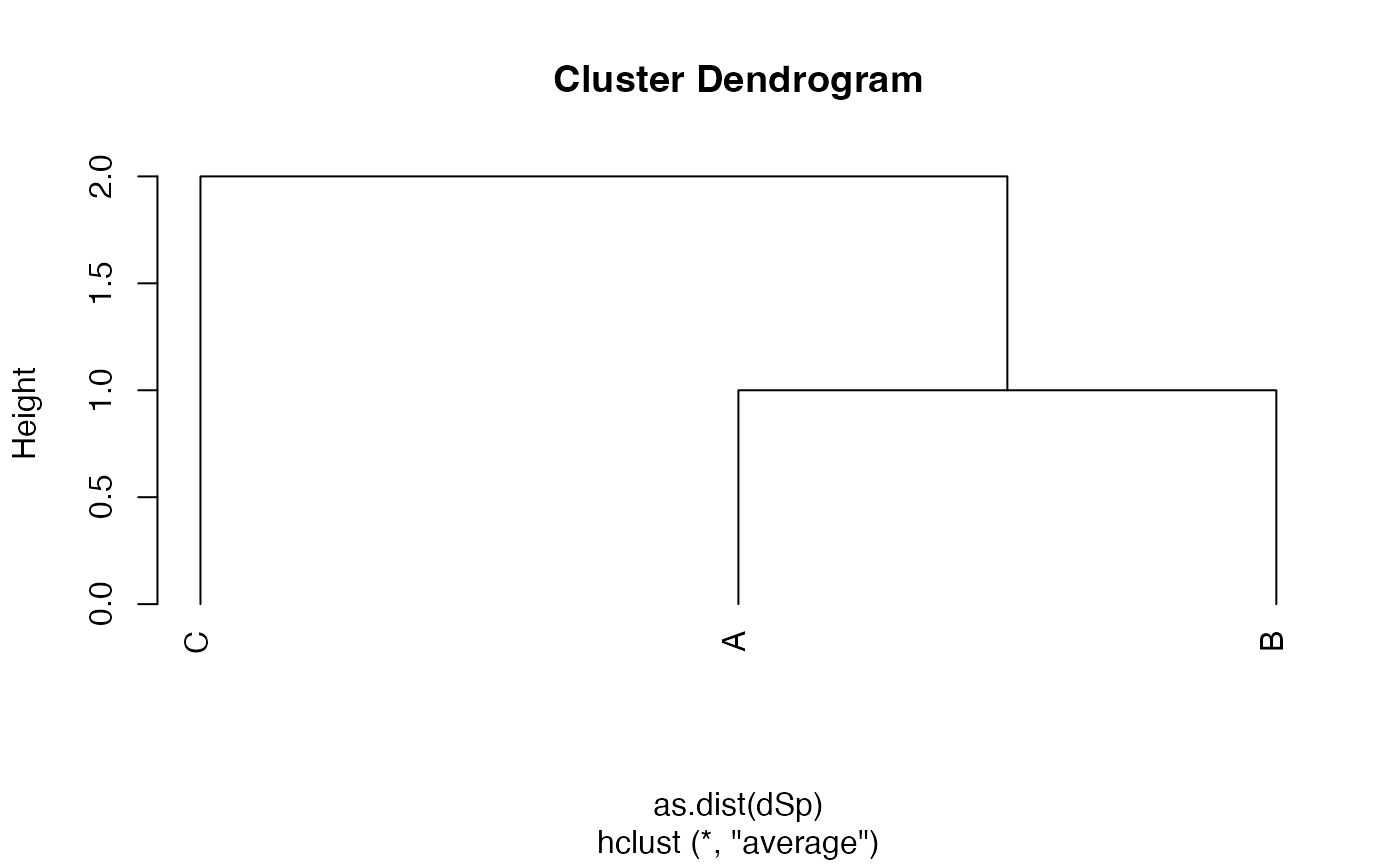
Node heights are stored in $height.
hTree$height## [1] 1 2phylo
Conversion to a phylo object is straightforward.
## Loading required package: ape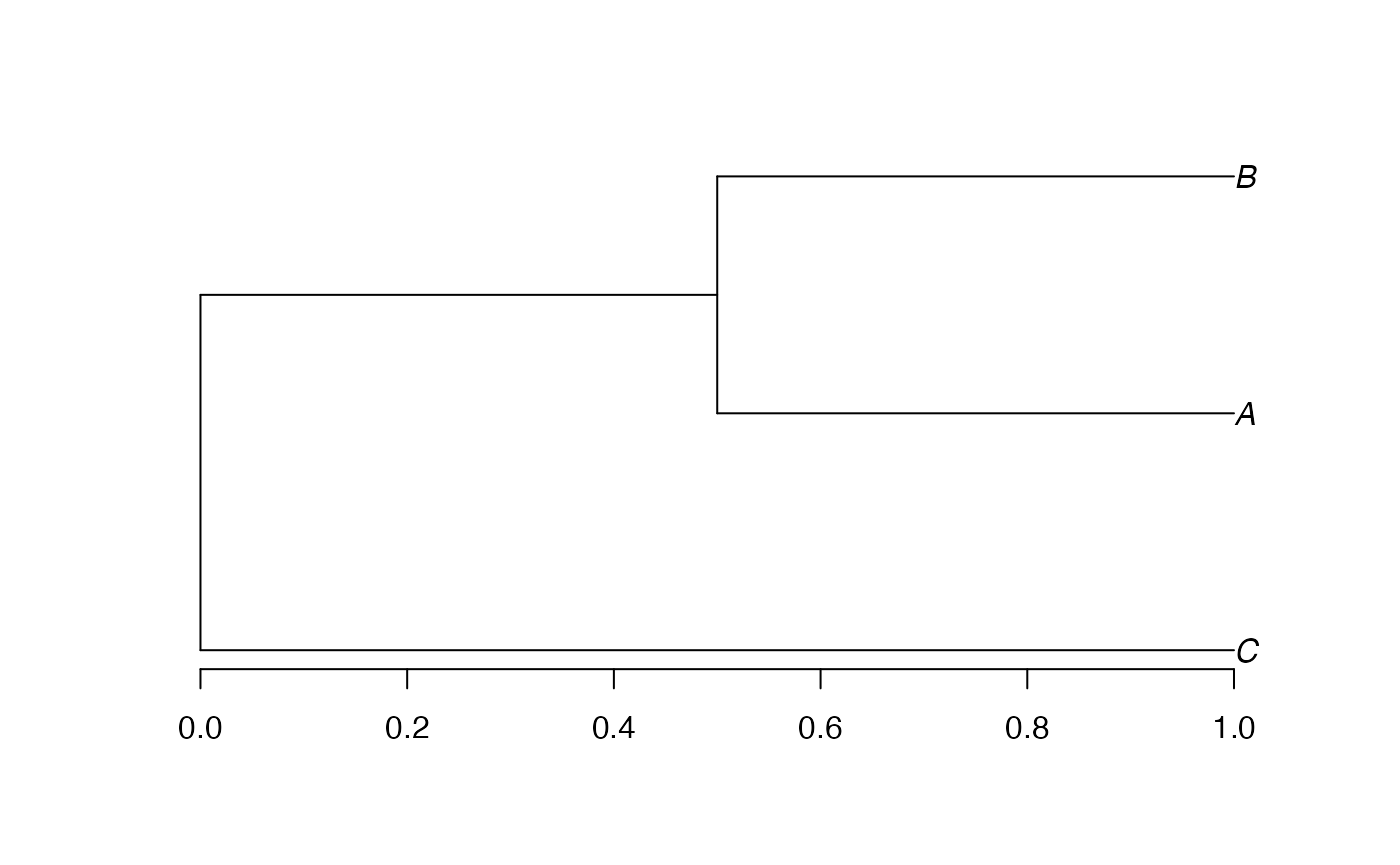
Edge lengths have been divided by 2 during the conversion, as
documented in ?as.phylo.hclust.
phyloTree$edge.length## [1] 1.0 0.5 0.5 0.5That does not suit our needs. In entropart, edge lengths are
multiplied by 2 after conversion, so that phyloobjects can
be identical to other phylogenies.
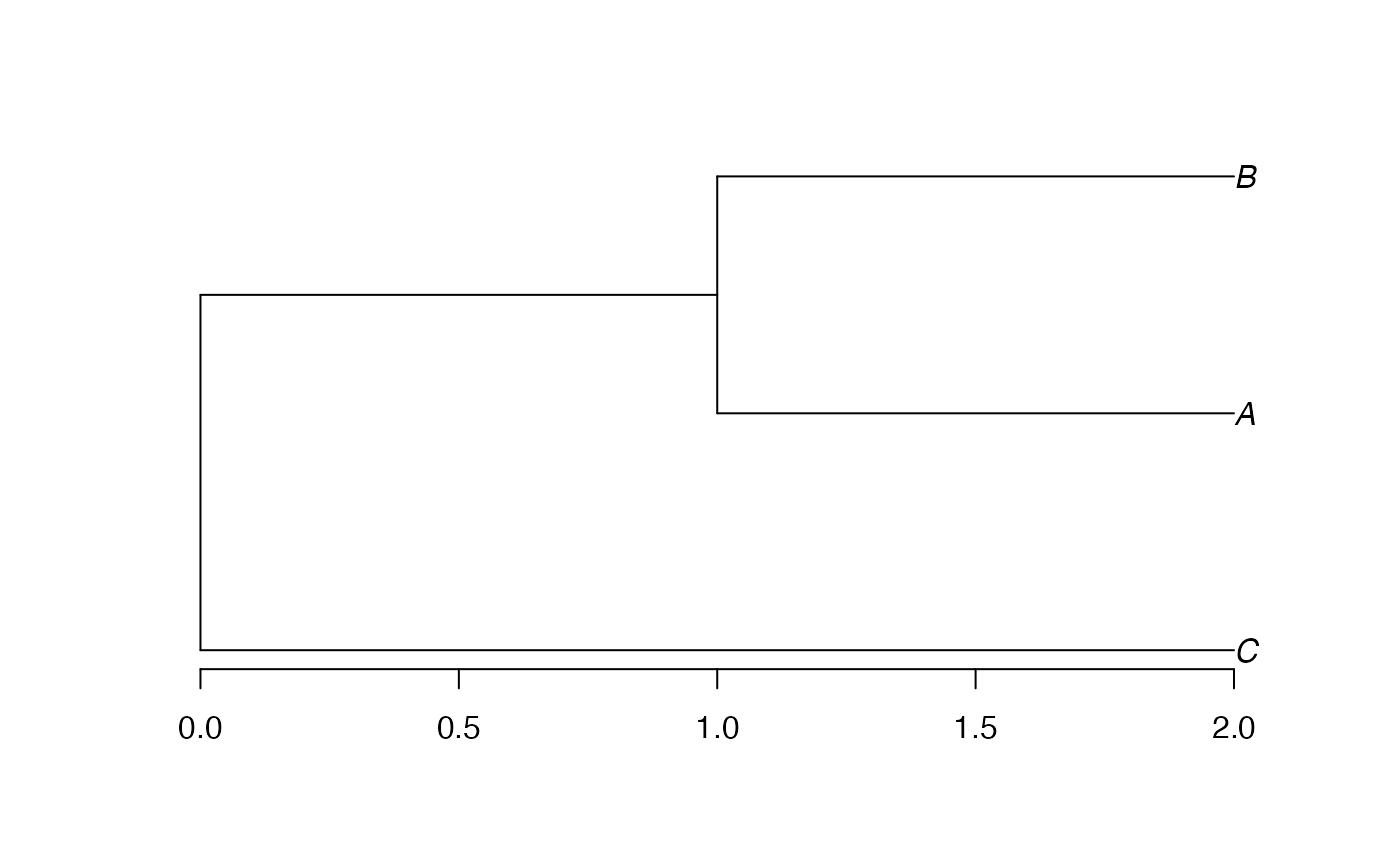
phylog
The last conversion is from phylo to
phylog.
## Loading required package: ade4
plot(phylogTree <- hclust2phylog(hTree))
axis(1)
Edge lengths are not stored directly in phylog objects.
The $droot component keeps distances from tips to root. The
$Wdist component is a dist (half a distance
matrix without the zero-diagonal) whose values are
,
where
is the distance between species
ant
.
phylogTree$droot## C A B Int1 Root
## 2 2 2 1 0
phylogTree$Wdist^2/2## C A
## A 2
## B 2 1