divent is a package for R designed to estimate diversity based on HCDT entropy or similarity-based entropy.
Several object classes can be used in the package to represent phylogenies and calculate phylogenetic diversity. They are detailed here.
Formats used
Supported phylogenies are classes phylo from package
ape, phylog from package ade4 and
hclust from package base.
To optimize performance, a specific class phylo_divent
is introduced by the package: it is basically a list containing a
phylo and an hclust representation of the
phylogeny and preprocessed useful statistics.
The argument tree used in phylogenetic diversity
estimation functions of the package may be any of those formats.
Conversion to phylo_divent is managed internally.
Conversion between classes
Let’s start from an ultrametric distance matrix. Three species are defined, with distance 1 between the first 2 and distance 2 between both and the last one.
species_dist <- matrix(c(0, 1, 2, 1, 0, 2, 2, 2, 0), nrow = 3, byrow = TRUE)
row.names(species_dist) <- colnames(species_dist) <- c("A", "B", "C")
species_dist## A B C
## A 0 1 2
## B 1 0 2
## C 2 2 0hclust
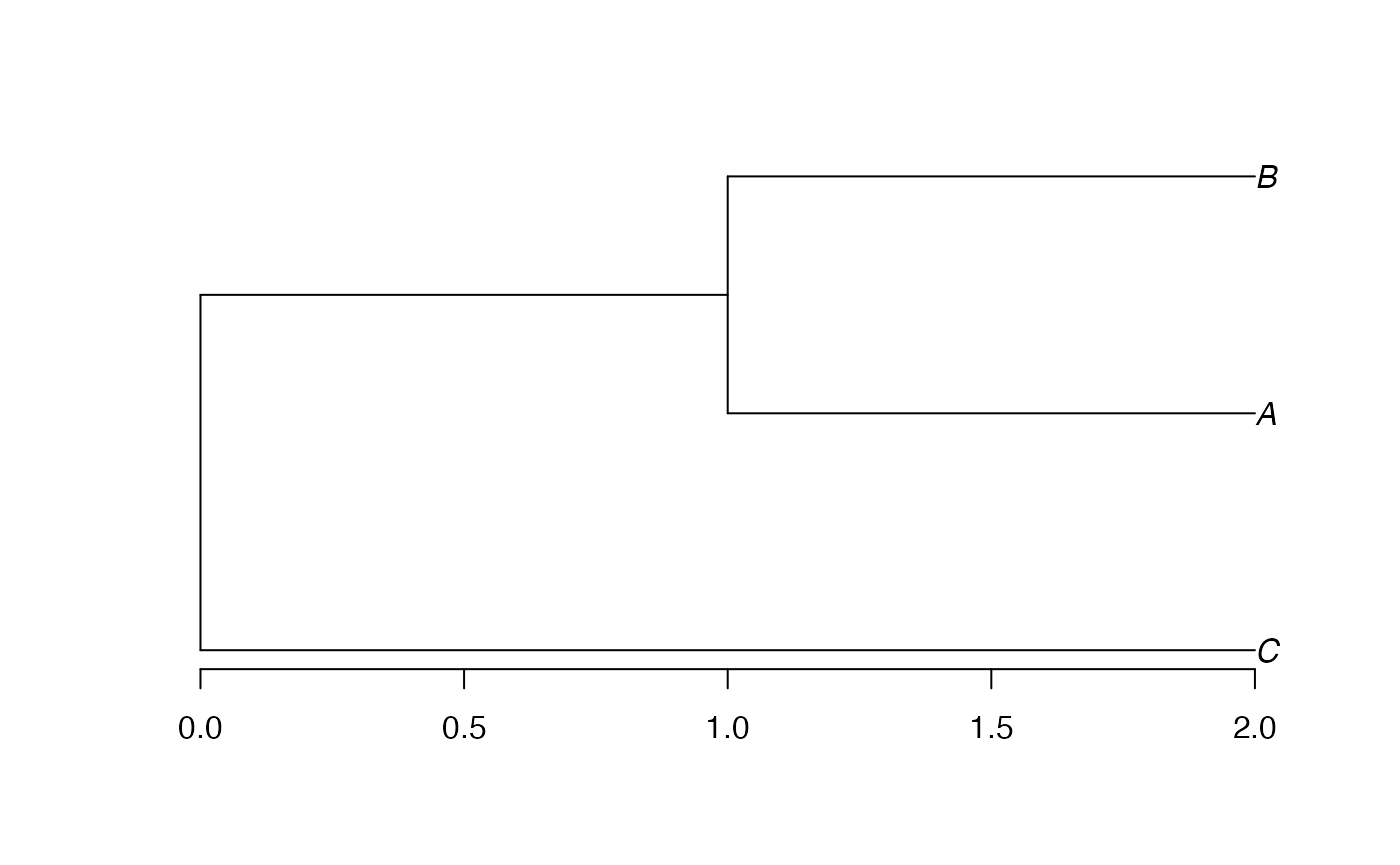
An hclust object is created by UPGMA hierarchical
clustering.
library("stats")
plot(
tree.hclust <- hclust(
as.dist(species_dist),
method = "average"
),
hang = -0.01,
axes = F
)
axis(2)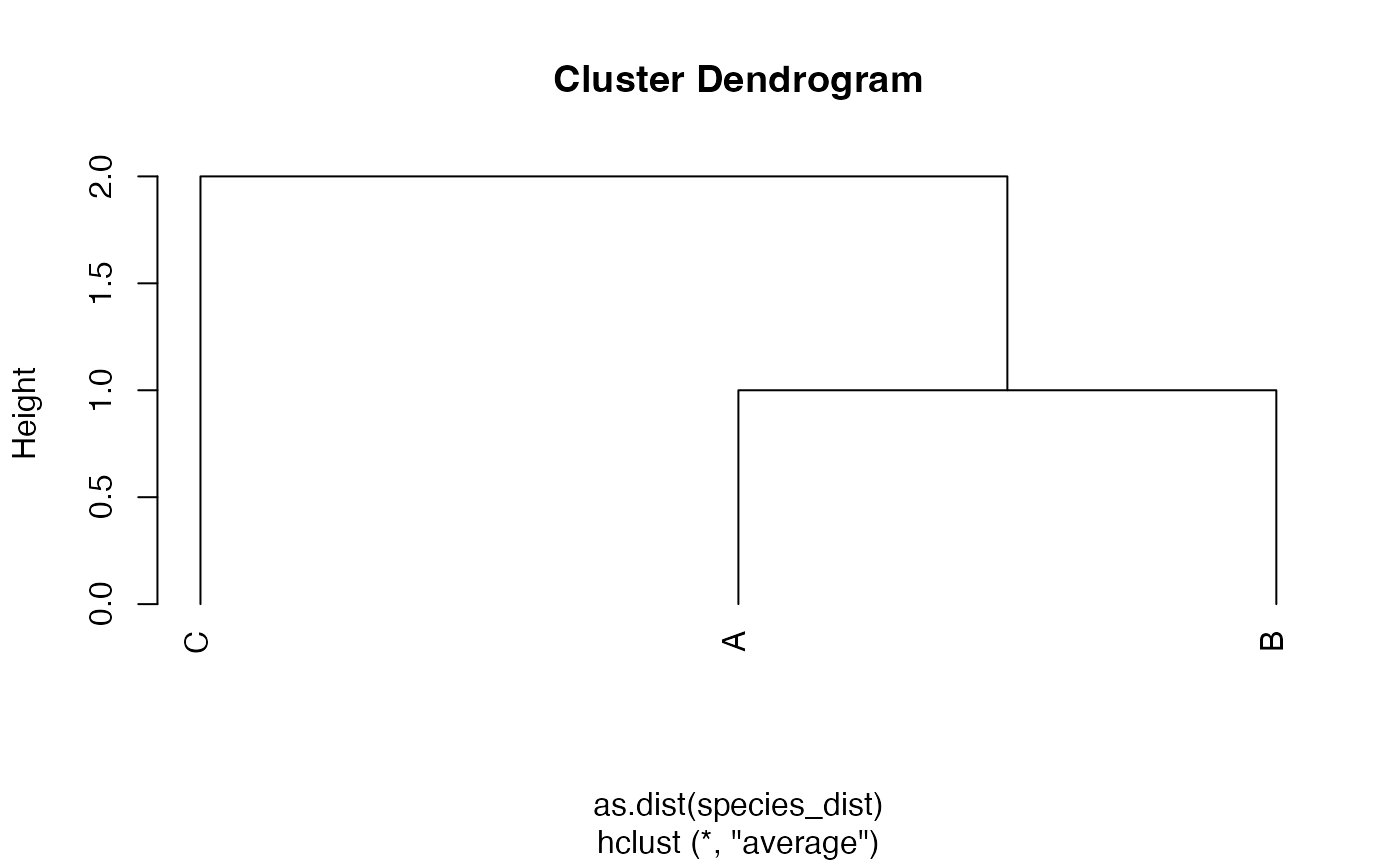
Node heights are stored in $height.
tree.hclust$height## [1] 1 2phylo
Conversion to a phylo object is straightforward.
Edge lengths have been divided by 2 during the conversion, as
documented in ?as.phylo.hclust.
tree.phylo$edge.length## [1] 1.0 0.5 0.5 0.5That does not suit our needs. In divent, edge lengths are
multiplied by 2 after conversion, so that phylo objects can
be identical to other phylogenies.
phylog
The last conversion is from phylo to
phylog.
library("ade4")
plot(tree.phylog <- hclust2phylog(tree.hclust))
axis(1)
Edge lengths are not stored directly in phylog objects.
The $droot component keeps distances from tips to root. The
$Wdist component is a dist (half a distance
matrix without the zero-diagonal) whose values are \(d_{s,t}=\sqrt{2 \delta_{s,t}^2}\), where
\(\delta_{s,t}\) is the distance
between species \(s\) ant \(t\).
tree.phylog$droot## C A B Int1 Root
## 2 2 2 1 0
tree.phylog$Wdist^2/2## C A
## A 2
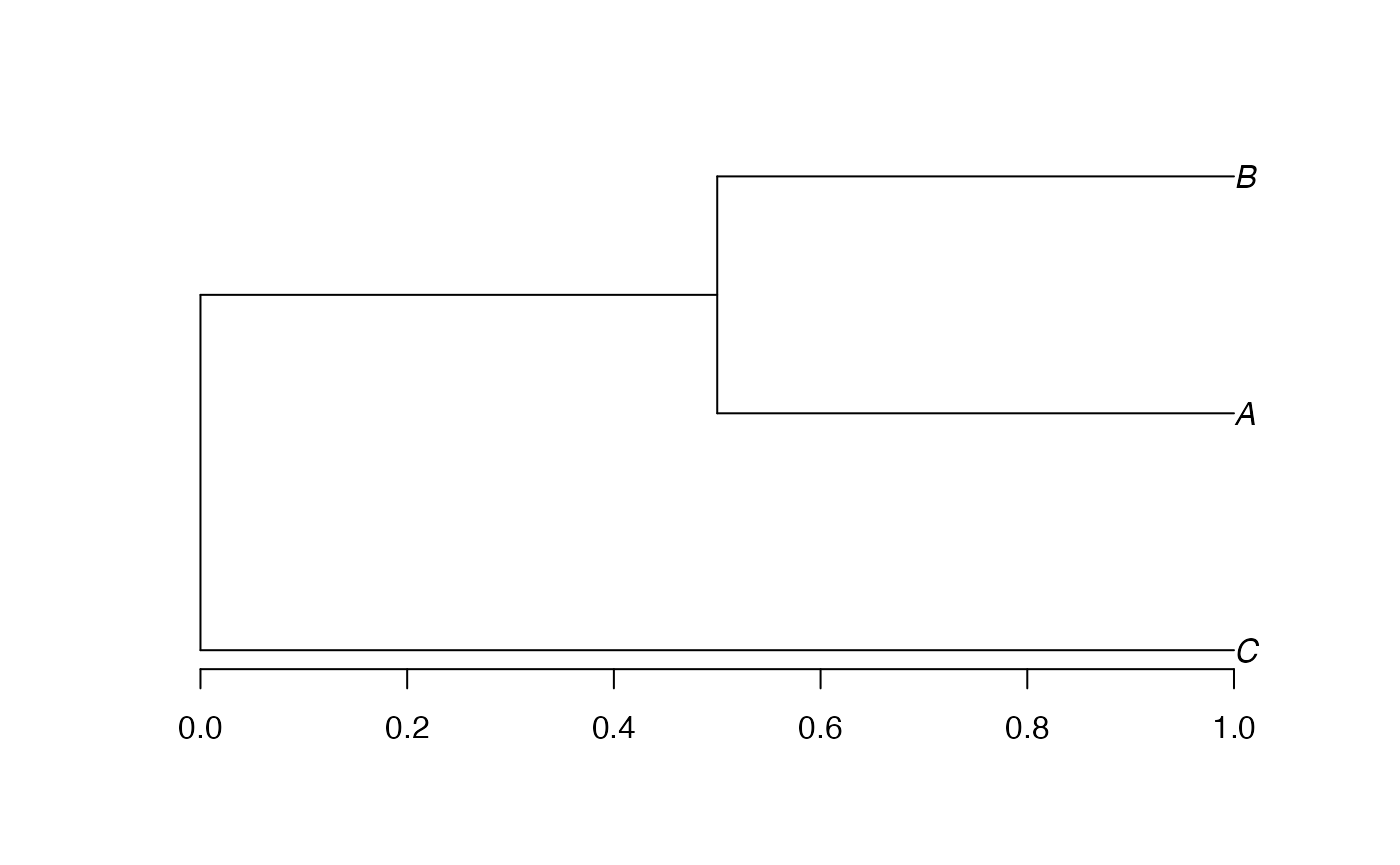
## B 2 1phylog trees are supported by divent but are
converted to hclust:
tree2.hclust <- stats::hclust(tree.phylog$Wdist^2 / 2, "average")
plot(tree2.hclust, hang = -0.01, axes = F)
axis(2)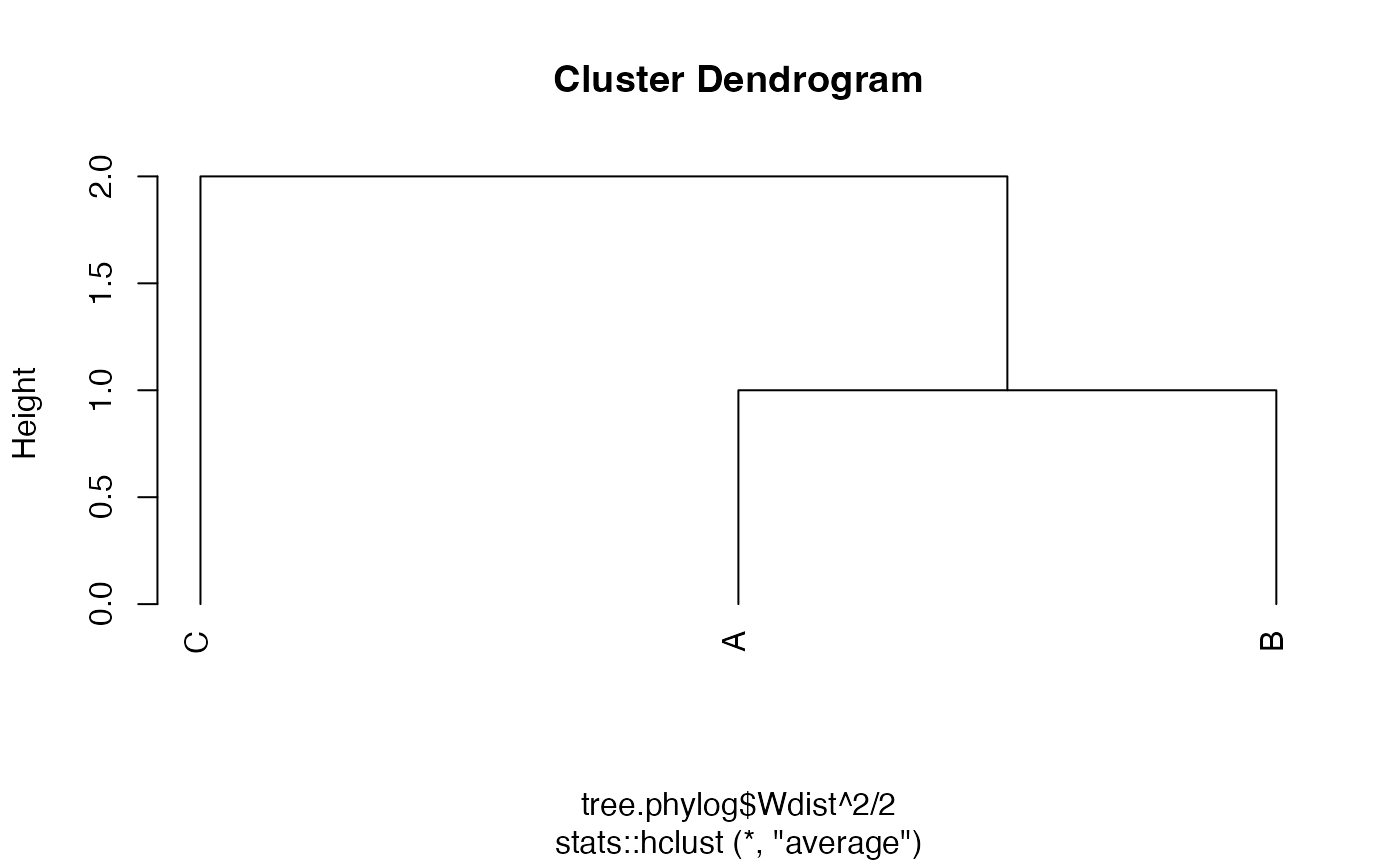
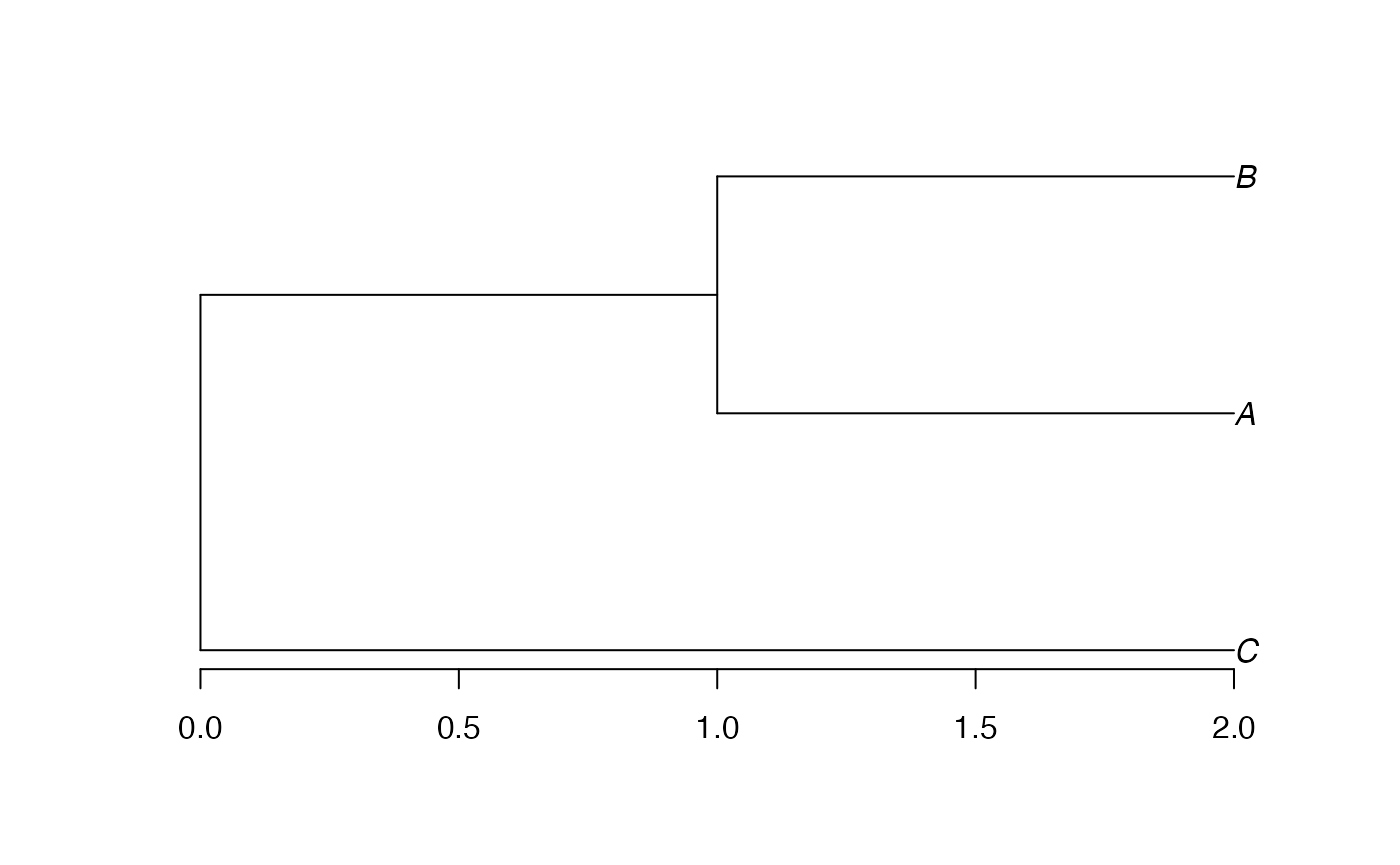
phylo_divent
The function as_phylo_divent converts any object of one
of the previous types.
## Loading required package: Rcpp
plot(tree.phylo_divent <- as_phylo_divent(tree.phylo))
axis(2)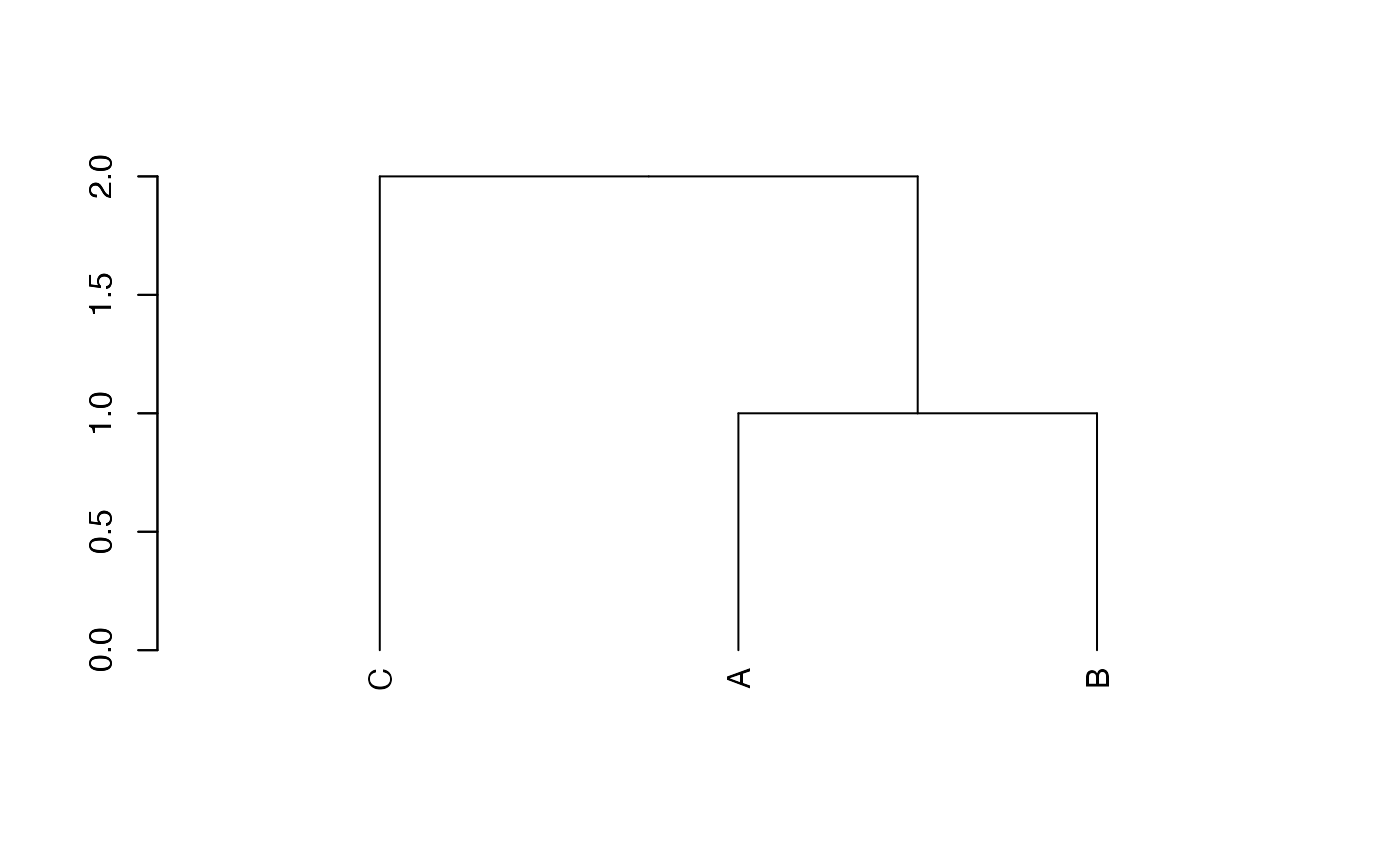
Its plot method is that of dendrogram
objects.
A phylo_divent tree is a list.
tree.phylo_divent## $phylo
##
## Phylogenetic tree with 3 tips and 2 internal nodes.
##
## Tip labels:
## A, B, C
##
## Rooted; includes branch length(s).
##
## $hclust
##
## Call:
## ape::as.hclust.phylo(x = tree)
##
## Cluster method : unknown
## Number of objects: 3
##
##
## $height
## [1] 2
##
## $cuts
## [1] 1 2
##
## $intervals
## [1] 1 1
##
## $phylo_groups
## 0 1
## A 1 1
## B 2 1
## C 3 2
##
## attr(,"class")
## [1] "phylo_divent"It contains:
-
$phylo: the tree as aphyloobject. -
$hclust: the tree as ahclustobject. -
$height: the height of the tree. -
$cuts: the heights of the nodes of the tree. -
$intervals: the intervals between the cuts. -
$phylo_groups: a matrix that describes the ancestors of species in each interval. The column names give the most recent cut of each interval. Values are the group each present species belongs to in each interval. In the example above, species A and B belong to the same group in the interval starting at 1.